



Статьи
Редкие нарушения липидного обмена — ситостеролемия
Ситостеролемия входит в группу рецессивных наследственных заболеваний. Измененные мутациями гены, кодирующие АТФ-связывающие белки-полутранспортеры, нарушают метаболизм стеринов, дезорганизуя процессы их поглощения и удаления из плазмы. В результате возникает дислипидемия стеролового генеза, повышающая риск развития атеросклероза. Пациентам нужна специализированная диета с низким содержанием растительных жиров, а также терапия эзетимибом и холестирамином.
ЕВРОПЕЙСКАЯ МУТАЦИЯ
Ситостеролемия (ССЛ), или болезнь накопления растительных эфиров, представляет собой редкую генетически обусловленную дислипидемию, наследующуюся по аутосомно-рецессивному типу. В ее патогенезе главную роль играют кишечная гиперабсорбция растительных стеринов, их пониженная билиарная экскреция, а также накопление в плазме и тканях организма.
Пока нет точных данных о распространенности ССЛ, однако известно, что мутация в гене ABCG8 встречается преимущественно у представителей европеоидной расы (немцы, чехи, индийцы, индейцы, закрытые религиозные группы, в том числе гуттериты и амиши), тогда как мутация ABCG5 чаще регистрируется у китайцев и японцев. Распространенность ССЛ в популяции составляет 1 случай на 2,6 млн человек с выявленной мутацией в гене ABCG5 и 1 на 360 тыс.— в ABCG8. В России в 2012 году заболевание встречалось в одном случае на 1 млн СЛЛ обнаруживается с равной частотой у обоих полов и не зависит от возраста. Так, например, случай ССЛ с ксантоматозом описан у ребенка 11 месяцев.
Как уже говорилось выше, основная причина недуга — биаллельная мутация в генах ABCG5 или ABCG8, кодирующих АТФ-связывающие белки-полутранспортеры, которые локализованы у человека главным образом на гепатоцитах и энтероцитах проксимального отдела тонкой кишки — стеролин-1 и -2 (STSL1 и STSL2). Нормальная функция STSL1 и STSL2, образующих между собой особый комплекс — гетеродимер, способствует обратному захвату холестерина и растительных стеролов и их экскреции в просвет кишки, с одной стороны, и стимулирует их печеночную экскрецию в гепатоцитах — с другой, осуществляя так называемый небилиарный путь выведения стеринов.
В здоровом организме фитостеролы (ситостерол, сигмастерол и кампестерол) повышают активность нормально функционирующих холестерин-связывающих белков, тем самым снижая всасывание холестерина и уменьшая его поступление в кровоток. Дефектные полутранспортеры неспособны адекватно выполнять свои функции, что неизбежно ведет к токсичному накоплению растительных стеринов и холестерина липопротеинов низкой плотности (ХС ЛПНП), а значит, к высокому риску развития атеросклероза.
Повышение концентрации общего холестерина до 17 ммоль/л — основной лабораторный признак, позволяющий заподозрить ССЛ, но, по некоторым данным, уровень общего холестерина у больных ситостеролемией может быть нормальным или слегка повышенным. В одном исследовании выявлена корреляция между высокими цифрами липидного профиля и наличием ксантом у детей и подростков.
 АЛГОРИТМ ДИАГНОСТИКИ
АЛГОРИТМ ДИАГНОСТИКИ
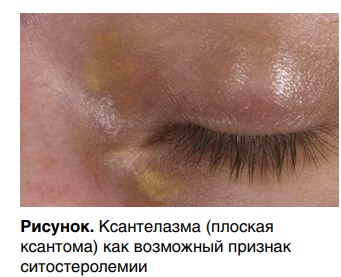
Клинические проявления болезни весьма различны и, как правило, представлены кожными, сухожильными или тубероэруптивными ксантомами и ксантелазмами (рис.), а в некоторых случаях—спленомегалией, артралгиями и артритами.
В литературе представлен единственный случай печеночной недостаточности, которая привела к циррозу печени и, вероятно, была следствием ССЛ у пациента с отрицательными маркерами гепатитов и других причинных факторов, которые могли бы запустить данные изменения.
Следует отметить, что диагностика ССЛ затруднена ввиду схожести клиниколабораторных проявлений у детей с симптомами гомозиготной формы семейной гиперхолестеринемии (гоСХГС). Зачастую это приводит к постановке ошибочного диагноза. Как показывает практика, ССЛ может быть заподозрена при отсутствии ответа на этапе терапии статинами, когда стандартные дозы препарата не оказывают клинического эффекта. В случае ССЛ статины не могут быть эффективными, поскольку их терапевтической мишенью является снижение синтеза холестерина за счет подавления активности ГМК-Коа-редуктазы — фермента, играющего ключевую роль в данном процессе.
Бразильские исследователи провели скрининг среди пациентов с гиперхолестеринемией и отрицательными результатами генетического исследования на ее семейную форму. У 3,1 % обследуемых были выявлены мутации генов ABCG5 и ABCG8, ответственных за развитие ССЛ. Применение каскадного скрининга позволило выявить шесть новых случаев заболевания и 18 гетерозиготных носителей.
Согласно данным крупного американского исследования, около 4 % пациентов с гиперхолестеринемией (уровень ЛПНП в крови выше 10,5 ммоль/л) параллельно имели повышенный В-ситостерол, тогда как у 0,3 % отмечалась явная ССЛ.
Не стоит забывать, что, помимо гоСГХС, в дифференциально-диагностический алгоритм должны быть включены такие заболевания, как дефицит лецитин-холестерин-ацилтрансферазы, болезнь Танжера, церебротендинозный ксантоматоз, семейная дисбеталипопротеинемия, семейная гипертриглицеридемия.
Патогномоничны для ССЛ изменения в гемограмме, характерные для гемолитической анемии и макротромбоцитопении. Они регистрируются в 25–35 % случаев. Так, в мазке периферической крови выявляются гигантские тромбоциты и стоматоциты. Данные изменения могут объясняться включением растительных стеролов в мембраны эритроцитов и тромбоцитов, которые приводят к их гиперактивации, повышая степень гидратации и вызывая осмотическую нестабильность.
Гематологические аномалии, которые могут быть единственным признаком ССЛ, зачастую трактуются как проявление иммунной тромбоцитопении, в результате чего больные рискуют получать ненадлежащую и бесполезную терапию. При ССЛ концентрация растительных стеринов может повышаться в сыворотке крови более чем в 10–25 раз, притом что нормальные значения не должны превышать 10–15 мг/л. Стойкие лабораторные изменения, характерные для ССЛ, отмечаются у гомозиготных пациентов, тогда как у носителей одного патогенного варианта может не наблюдаться патологического повышения фитостеролов в крови.
МЕТОДЫ ЛЕЧЕНИЯ
Главный элемент немедикаментозного лечения ситостеролемии — диетотерапия, основанная на ограничении потребления природных и промышленных продуктов, содержащих фитостерины.
Пациенту следует воздержаться от потребления орехов, семечек, бобовых, зародышей пшеницы, оливок, авокадо, моллюсков, водорослей, растительных масел, шоколада. Рекомендуется исключить маргарин, спреды, шортенинг. Вместо цельного зерна лучше отдавать предпочтение обработанному рису. Тем не менее полностью исключить поступление фитостеринов в организм невозможно, так как они входят в состав всех продуктов растительного происхождения, а соблюдение диетических рекомендаций помогает снизить уровень растительных стеролов не более чем на 30 %. Поэтому для достижения наилучшего эффекта контроль в питании сочетают с медикаментозной терапией.
«Золотым стандартом» фармакотерапии ССЛ признан эзетемиб, способный ингибировать белок Niemann-Pick C1-Like1 (NPC1L1), отвечающий за первичное всасывание холестерина и растительных стеролов. Препарат оказывает влияние на уровне кишечника, при этом сам он не всасывается, но подавляет всасывание стеринов. По мере уменьшения уровня циркулирующих фитостеринов на фоне терапии эзетимибом наблюдается увеличение размера и количества тромбоцитов. Терапия данным препаратом безопасна и эффективно используется в педиатрической практике.
При недостаточном ответе на ингибитор NPC1L1 можно использовать секвестранты желчных кислот (ЖК), обычно это холестирамин, который, связывая ЖК, ингибирует их реабсорбцию в подвздошной кишке. В наиболее тяжелых случаях прибегают к ее частичному шунтированию.
ССЛ часто остается нераспознанной, особенно при гетерозиготном варианте заболевания, когда клиническая картина долго сохраняется латентной. Разработанные рекомендации предлагают проведение универсального скрининга детей в 9–11 лет и затем повторно — в 17–21 год для выявления новых случаев гиперхолестеринемии. Данная программа поможет идентифицировать различные виды дислипидемий, что станет первым этапом для дальнейшей диагностики СЛЛ.
Особое внимание нужно уделять пациентам с выраженной гиперхолестеринемией без мутации генов, ответственных за развитие семейной гиперхолестеринемии.
Список литературы находится в редакции
Читайте также
- Пробиотики в терапии аллергической патологии у детей
- Глютензависимые заболевания: классификация и основы патогенеза
- Современные технологии мониторинга углеводного обмена у детей с сахарным диабетом 1 типа
- Значение генетического исследования церебрального паралича
- Менингококковая инфекция: современные вызовы и решения
- Лечение инфекций мочевыводящих путей
НАШИ ПАРТНЕРЫ















