



Статьи
Цифровой фенотип: современная информационно-аналитическая платформа для выявления новых гено-фенотипических связей
Уникальная информационно-аналитическая платформа «Цифровой фенотип» (ЦФ) разработана на основе сочетанного применения экспертного глубокого фенотипирования ряда редких наследственных инвалидизирующих заболеваний, манифестирующих в детском возрасте, и современных информационных технологий для построения веб-приложений.
Глубокое фенотипирование подразумевает формализованное и максимально полное описание совокупности клинического проявления каждого наследственного заболевания. Это имеет важное значение для анализа и выявления устойчивых гено-фенотипических связей с целью повышения надежности диагностики и прогнозирования характера течения наследственных заболеваний.
Платформа ЦФ позволяет:
1) осуществлять ввод формализованных клинических характеристик с оценкой степени выраженности фенотипических признаков;
2) вводить результаты генетических исследований;
3) формировать поисковые запросы для экспорта в Excel выборочных цифровых данных для последующего анализа.
ПРОБЛЕМА РЕДКИХ БОЛЕЗНЕЙ
Пациенты с редкими заболеваниями (а их насчитывается более 8 тысяч)—это сложная проблема для общества и системы здравоохранения. Хотя болезни и редкие, в целом ими страдают около 7 % населения Земли. По данным Всероссийского общества орфанных заболеваний, в России насчитывается более 2 миллионов таких больных. Налицо парадокс: болезни редки, а пациенты — многочисленны!
Дифференциальная диагностика редких наследственных заболеваний, манифестирующих в детстве, во многих случаях крайне сложна из-за многообразия нозологических форм при возможном и даже частом совпадении ряда фенотипических признаков, а также низкой распространенности и высокой вероятности сочетанной патологии при недостаточном клиническом опыте врачей. Несвоевременные диагностика и лечение детей с орфанными заболеваниями приводят к развитию множества осложнений, ранней инвалидизации, а зачастую и к преждевременной смерти. К сожалению, около трети детей с редкими заболеваниями не доживают до 5-летнего возраста.
Причины наследственных заболеваний кроются в генетических нарушениях, для выявления которых эффективно используются современные методы молекулярной биологии, такие как высокопроизводительное секвенирование и биоинформатический анализ. Создание баз данных нескольких сотен и тысяч пациентов детского возраста, содержащих результаты генетических исследований, позволяет на совершенно новом для медицинских исследований уровне выявлять взаимосвязи генетических изменений с фенотипическими проявлениями наследственных заболеваний.
Установление гено-фенотипических взаимосвязей дает возможность более эффективно решать вопросы генетического скрининга и предоставляет существенные клинические преимущества, позволяя выявлять генетические варианты, ассоциирующиеся с тяжелым течением болезни. Обнаружение новых гено-фенотипических связей служит основой для прогнозирования ухудшения состояния пациента и назначения персонифицированной терапии, в том числе больным с выявленными каузативными мутациями, у которых тяжелые последствия еще не наступили. Важно последовательно формировать массивы данных, опираясь на формализованные экспертные описания клинических проявлений орфанных заболеваний, для последующего анализа временных рядов и выявления гено-фенотипических корреляций.
РЕАЛИЗАЦИЯ ПРОЕКТА
В НИКИ педиатрии и детской хирургии имени академика Ю.Е. Вельтищева по инициативе его научного руководителя профессора М.А. Школьниковой и академика К.Г. Скрябина в 2018 году были начаты работы по созданию информационно-аналитической платформы «Цифровой фенотип». Были разработаны уникальные экспертные шкалы для глубокого фенотипирования целого ряда наследственных заболеваний, манифестирующих в детском возрасте, включая эпилептические энцефалопатии, первичные цилиарные дискинезии, первичны электрические заболевания сердца, синдромы Марфана, Ангельмана, Ретта.
Принцип глубокого фенотипирования, включающий (в зависимости от вида патологии) описание сотен формализованных признаков оценки пациента, был успешно реализован благодаря высокому уровню экспертизы. При этом описание степени выраженности фенотипических признаков пациентов формировалось с использованием номинальных, порядковых и количественных шкал, что позволило оценить признаки с учетом степени их выраженности и объективизировать последующий анализ любыми другими количественными характеристиками.
Не секрет, что именно качественное, а не количественное описание фенотипа служило препятствием к такому анализу. Именно детализированная формализация фенотипа на основании коллективного экспертного мнения в сочетании с математическим инструментом анализа позволила сделать большой шаг вперед в изучении наследственных заболеваний.
Для глубокого фенотипирования также были успешно использованы цифровые данные многих функциональных методов исследования, таких как электрокардиография и холтеровское мониторирование, электроэнцефалография головного мозга. Ранее результаты этих исследований вводились в электронную историю болезни и применялись специалистами для оценки динамики состояния пациента только в виде текстовых заключений. Таким образом, ценная информация о больших субпопуляциях пациентов не могла быть использована для научного анализа. Исключение представляли только диссертационные исследования, охватывающие небольшие группы больных.
Институт Вельтищева является ведущим лечебным учреждением федерального уровня, в которое направляют детей из разных регионов России с редкими заболеваниями, сложными с точки зрения постановки диагноза. Каждый год здесь проходят стационарное лечение от 9 до 14 тысяч детей, сведения о которых фиксируются в автоматизированной истории болезни, доступной врачам, которые участвуют в проекте ЦФ. Структурная схема информационно-аналитической платформы (ИАП ЦФ) представлена на рисунке 1.
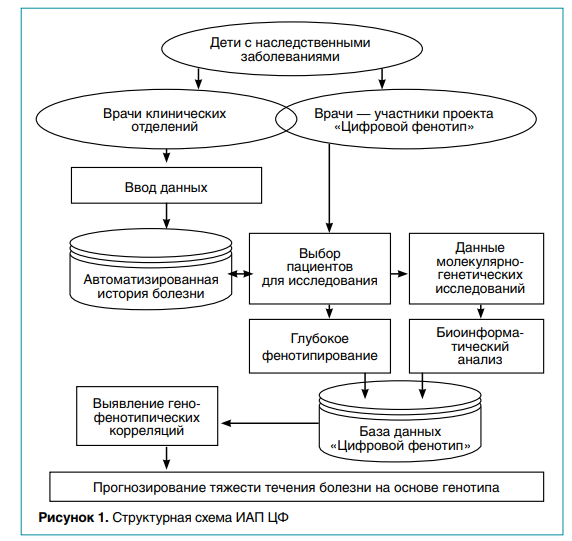
ИАП ЦФ, разработанная на основе современных информационных технологий для построения веб-приложений, позволяет осуществлять ввод и проверку корректности вводимых данных, оценку степени выраженности фенотипических признаков, формирование поискового запроса, вывод выборочных данных на экран и экспорт в Excel. В основе технологии проектирования программы ЦФ (свидетельство Роспатента о регистрации программы для ЭВМ № 2021666837 от 20.10.2021) лежит архитектура реляционной модели данных с использованием кроссплатформенного решения на базе стека Laravel — AngularJS — mySQL. Экранные формы структурированы для удобства визуализации, предусмотрены также динамические формы для ввода данных об имеющейся патологии у ближайших родственников ребенка (отец, мать, сибсы).
В процессе наблюдения за пациентом база пополняется визитозависимыми данными, включая вычисленные отклонения от нормы параметров физического развития ребенка с использованием центильных таблиц. Это позволяет анализировать динамику состояния пациентов, разрабатывать и верифицировать математические модели прогнозирования тяжести течения изучаемых заболеваний. Платформа ЦФ размещена на веб-сервере института и функционирует в локальной сети. Универсальность веб-интерфейса обеспечивает корректную работу при использовании браузеров Google Chrome, Opera, Яндекс, Safari, Edge, Firefox.
При работе с платформой врач, участник проекта, выбирает пациента для пополнения базы данных ЦФ по определенной нозологии. Интеграция с автоматизированной электронной историей болезни позволяет использовать ранее введенную структурированную информацию о больном, включая антропометрию, данные физикальных обследований, результаты инструментальных и лабораторных исследований, сформировав формализованное представление семейного анамнеза (рис. 2).
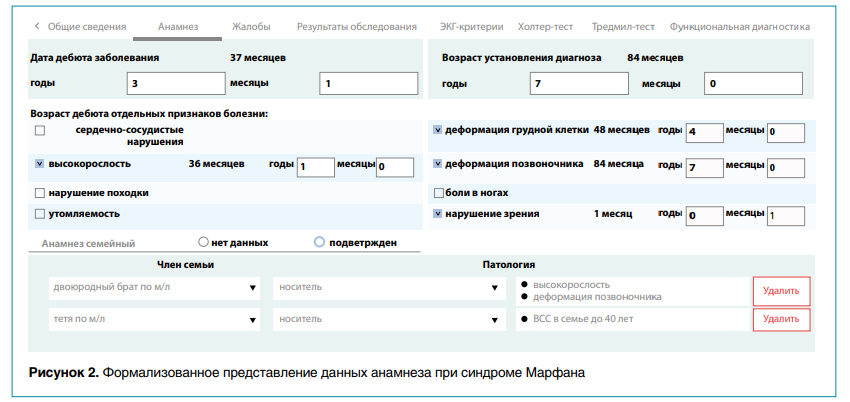
Затем осуществляется ввод фенотипических признаков по ранее разработанной шкале цифрового фенотипа для конкретного наследственного заболевания. Результаты молекулярно-генетических исследований при их наличии вводятся сразу или по мере их получения после биоинформатического анализа (рис. 3).

ПЕРВЫЕ РЕЗУЛЬТАТЫ
Развитие большинства наследственных форм патологии обусловлено генными мутациями. Нонсенс-мутации, мутации со сдвигом рамки считывания, мутации сайтов сплайсинга, делеции нескольких экзонов и другие перестройки ведут к нарушению синтеза соответствующего белка и потере его функции — Loss of Function (LoF). Миссенс-мутации не нарушают процесс синтеза белка, но оказывают негативный эффект на молекулярные процессы в клетках, нередко вызывая фенотипические проявления болезни.
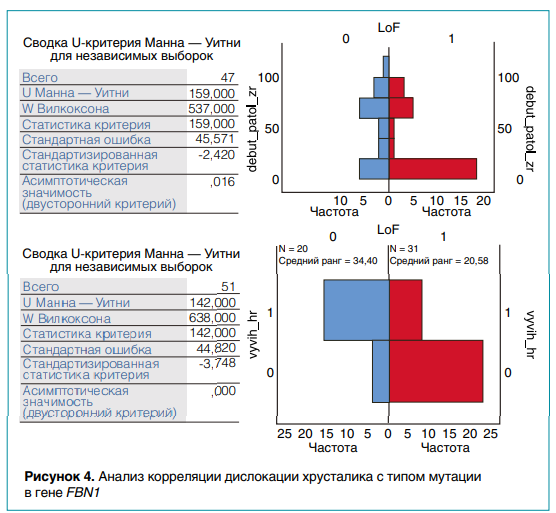
Цель гено-фенотипического анализа заключалась в выявлении взаимосвязи фенотипических проявлений моногенных наследственных заболеваний и типа мутаций. Из базы данных ИАП ЦФ была выделена выборка из 51 пациента с генетически подтвержденным синдромом Марфана, включая 31 ребенка с генетическим дефектом, связанным с полной потерей функции белка (LoF = 1), а у 20 детей изменения в нуклеотидной последовательности ДНК были вызваны миссенс-мутацией (LoF = 0).
Для выявления зависимости сроков манифестации и тяжести течения болезни от типа мутаций для гено-фенотипического анализа был использован статистический пакет SPSS. В результате применения непараметрического критерия Манна—Уитни показано, что при LoF = 1 у детей с синдромом Марфана патология органов зрения (миопия, дислокация хрусталика) манифестирует раньше (уровень значимости = 0,016), чем у больных с миссенс-вариантами. В то же время вывих или подвывих хрусталика имеет место преимущественно у детей с миссенс-мутациями (уровень значимости = 0,001). При этом лишь у 20 % детей из данной группы не было такого признака (рис. 4). Полученные данные позволяют предсказывать раннюю манифестацию дислокации хрусталика у больных с синдромом Марфана с миссенс-мутациями гена FBN1.
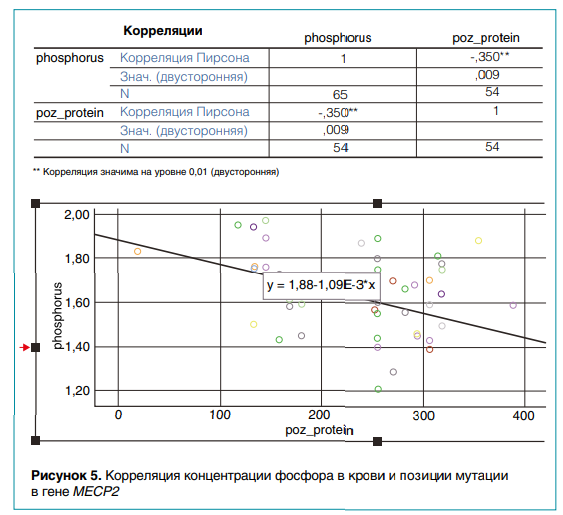
Результаты анализа корреляций клинических и генетических данных пациентов можно проиллюстрировать на примере другого моногенного наследственного заболевания—синдрома Ретта. Он встречается в основном у девочек и характеризуется регрессом психомоторного развития. Причина патологии — в мутации гена MECP2. Применение корреляционного анализа позволило выявить взаимосвязь фенотипического признака, концентрации неорганического фосфора в крови, с генетическим — позицией мутации в гене MECP2. Выявлена значимая отрицательная корреляция (r = –0,35; уровень значимости = 0,010) концентрации фосфора и позиции мутации (рис. 5).
ИАП ЦФ, разработанная на основе опыта ведущих экспертов в области наследственных заболеваний, позволяет реализовать принцип глубокого фенотипирования в виде формализованного описания нескольких сотен фенотипических признаков пациента, включая оценку степени их выраженности с использованием порядковых и количественных шкал.
Первые результаты выявления гено-фенотипических взаимосвязей на основе статистических методов анализа данных показали возможность их применения в клинической практике. Если у пациента с патологией соединительной ткани и вовлечением сердечно-сосудистой системы есть эктопия хрусталика, то можно с большой вероятностью предположить диагноз синдрома Марфана с миссенс-вариантом в гене FBN1. Обнаружено также, что у детей с мутациями на C-конце белка MECP2 уровень фосфатов в крови ниже, чем у больных с мутациями на N-конце белка. Данные об уровне неорганических фосфатов у детей с синдромом Ретта важны для понимания причин остеопороза и назначения терапии с целью предотвращения возможных переломов костей скелета. При этом отрицательная зависимость концентрации фосфатов от позиции мутации в гене MECP2 ранее не была описана.
ПЕРСПЕКТИВЫ РАЗВИТИЯ
Анализ корреляций клинических и генетических данных на основе ИАП ЦФ при наличии достаточного количества пациентов в базе данных позволит разрабатывать модели оценки тяжести течения заболевания с учетом генотипа больного, а также определять возможные молекулярные мишени для дифференцированной терапии. Статистический анализ формализованных фенотипических данных облегчает поиск наиболее вероятных генетических дефектов, позволяет обосновать целенаправленный поиск патогенных мутаций, избежав неоправданных временных и экономических потерь, а главное — потерь здоровья и качества жизни пациентов с наследственными заболеваниями.
Благодаря работе благотворительного фонда «Геном жизни» и расширению спектра генетических исследований появляется все больше пациентов с качественными результатами экзомного и полногеномного секвенирования по всей России, поэтому в дальнейшем мы предполагаем предоставить доступ к ИАП ЦФ врачам из разных территорий страны для ввода и анализа гено-фенотипических данных пациентов с орфанными заболеваниями на региональном уровне. В последующем, вероятно, под эгидой Минздрава, целесообразно формирование интегрированной базы данных по редким болезням, что позволит объединить усилия врачей-исследователей по анализу данных, включая разработку математических моделей прогнозирования тяжести течения заболевания в зависимости от генотипа больного
НАШИ ПАРТНЕРЫ















