



Статьи
Синдром Гурлер
Мукополисахаридоз 1‑го типа относится к наследственным нарушениям обмена. Это группа заболеваний, при которых из-за генетического дефекта блокируется процесс метаболизма, что приводит к накоплению метаболитов или дефициту продукции фермента. Большинство наследственных нарушений обмена веществ крайне редки по отдельности, но в целом они составляют довольно распространенную группу расстройств среди детей.
МУТАЦИИ В СТРУКТУРНОМ ГЕНЕ
Мукополисахаридоз (МПС) 1‑го типа наследуется по аутосомно-рецессивному типу. По разным данным, в популяции он встречается с частотой один случай на 40 000–100 000 новорожденных. Развитие заболевания связано с мутациями в структурном гене лизосомального фермента альфа-L-идуронидазы IDUA, что приводит к дефициту фермента и накоплению в тканях нерасщепленных продуктов обмена — гликозаминогликанов (ГАГ). Изменения практически во всех органах и тканях клинически проявляются поражением нервной системы, умственной отсталостью, сердечно-легочными нарушениями, гепатоспленомегалией, задержкой роста, деформациями скелета, множественным дизостозом, помутнением роговицы.
Выделяют три формы МПС 1‑го типа:
- синдром Гурлер (МПС I-H — тяжелая форма) встречается примерно у 50–80 % пациентов;
- синдром Гурлер — Шейе (МПС I-H/S — промежуточная форма);
- синдром Шейе (МПС I-S — легкая форма).
СТРАТЕГИИ ТЕРАПИИ
Сегодня широко применяются две стратегии терапии МПС 1-Н. Во-первых, консервативная фермент-заместительная терапия (ФЗТ), максимально продлевающая жизнь пациентов — до 6–12 лет. Этот относительно безопасный и эффективный метод демонстрирует улучшение функции легких и физической активности, снижение уровня ГАГ в тканях, уменьшение выраженности гепатоспленомегалии. Однако он не позволяет надеяться на сохранение нормальной функции центральной нервной системы (ЦНС), поскольку препарат не проникает через гематоэнцефалический барьер. Другая проблема ФЗТ — возможное снижение эффективности терапии ввиду иммуногенности препарата. Во-вторых, используется аллогенная трансплантация гемопоэтических стволовых клеток (алло-ТГСК), направленная на коррекцию дефекта восстановлением физиологических функций. Алло-ТГСК приводит к эндогенному обеспечению недостающим ферментом всех тканей и органов. Ее успех зависит от нескольких факторов, главные из которых — возраст ребенка на момент трансплантации и степень тяжести клинических проявлений заболевания. Сохранение когнитивных функций наблюдается у детей в возрасте до 24 месяцев с более высоким коэффициентом психомоторного развития, что дает наибольший эффект алло-ТГСК в данной группе пациентов. Долгосрочная выживаемость зависит также от выбора оптимального донора для обеспечения стойкого приживления с достижением полного донорского химеризма и от поддержания нормального уровня фермента после алло-ТГСК с минимальным риском развития реакции «трансплантат против хозяина» (РТПХ). Среди возможных вариантов рассматривается использование совместимого по генам HLA-системы родственного донора, не являющегося носителем, а также совместимого неродственного донора. При этом в создании иммунологической толерантности, необходимой как для приживления, так и для минимизации проявлений иммунологического конфликта, важную роль играют режимы кондиционирования (РК) и способы профилактики РТПХ. При выборе режима кондиционирования и его интенсивности у детей, тем более с наследственными заболеваниями, необходимо учитывать степень риска токсических осложнений.

СОБСТВЕННЫЙ ОПЫТ
В клинике НИИ ДОГиТ им. Р.М. Горбачевой ПСПбГМУ им. И.П. Павлова в период с апреля 2010 г. по февраль 2020 г. выполнены алло-ТГСК 28 пациентам с МПС 1-Н. Диагноз устанавливали на основании характерной клинической картины, критически низкого уровня альфа-L- идуронидазы, повышения уровня экскреции ГАГ в моче, молекулярно-генетически подтвержденного дефекта гена IDUA. Медиана возраста на момент постановки диагноза составила 15 (3–35) мес. В качестве терапии на этапе до трансплантации использовали ФЗТ ларонидазой в стандартном режиме — 100 Ед/кг еженедельно в виде внутривенной инфузии. Медиана возраста на начало ФЗТ — 20 (4–40) мес, длительности терапии — 5 (1–20) мес. Возраст пациентов на момент алло-ТГСК составлял от 10 до 44 месяцев, медиана — 24 мес. В большинстве случаев (27/28) алло-ТГСК выполняли от неродственного донора, как полностью (n = 23), так и частично (n = 4) совместимого по генам HLA-системы. В качестве РК использовали миелоаблативные схемы (МАК) (14/28) на основе бусульфана либо треосульфана, а также флударабин + мелфалан-содержащие РК со сниженной интенсивностью доз (РИК) (14/28). Основанием для применения РИК был более старший возраст пациентов на момент алло-ТГСК (медиана возраста в группе МАК составила 23 мес, в группе РИК — 28 мес), а также значимые осложнения основного заболевания, в частности острое нарушение мозгового кровообращения в анамнезе, кардиомиопатия и хронический миокардит, тяжелая пневмония смешанного генеза (по одному эпизоду) и др. Пятилетняя общая выживаемость (ОВ) в группе пациентов, получивших алло-ТГСК, составила 89 % (рис. 1а), бессобытийная выживаемость (БСВ) — 57 %. Приживление трансплантата достигнуто у 27/28 пациентов. Медиана восстановления лейкоцитов до >1,0×109/л составила 19 (12–33) дней, нейтрофилов до >0,5×109/л — 23 (12–33) дня. У 1 пациента зарегистрировано первичное неприживление трансплантата после алло-ТГСК от полностью HLA-совместимого неродственного донора с МАК (Bu12/ Flu150). Возраст этого пациента на момент выполнения алло-ТГСК составил 44 месяца. Пациенту выполнена повторная алло-ТГСК от того же донора с достижением восстановления показателей периферической крови и полного донорского химеризма.
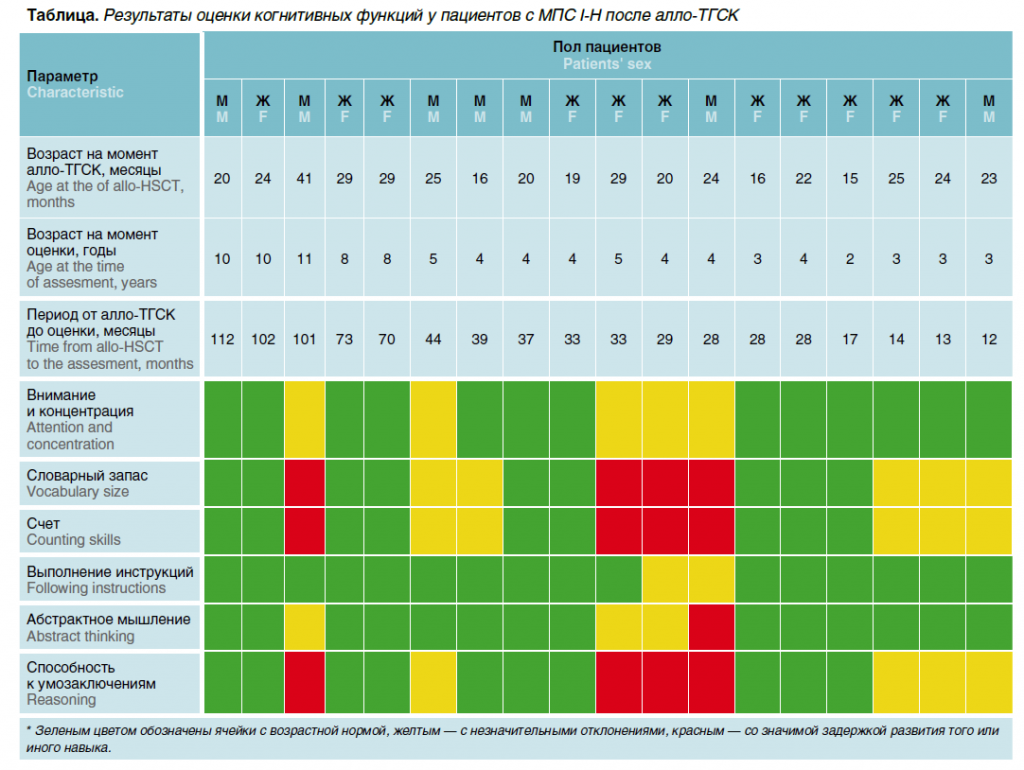
Вторичное отторжение трансплантата в отдаленном периоде алло-ТГСК после имевшегося приживления зарегистрировано у 4 пациентов. Все они получили повторную алло-ТГСК без смены донора, приживление достигнуто во всех случаях, однако у 1 пациента на Д+105 зарегистрировано повторное отторжение трансплантата, в связи с чем возобновлена ФЗТ. Острая РТПХ II–IV степени тяжести зарегистрирована у 12, III–IV степени — у 5 пациентов. Таким образом, кумулятивная частота острой РТПХ II–IV степени составила 43 %, III–IV степени — 18 %. Фактором, значимо снижающим риски развития данного осложнения, стало использование посттрансплантационного циклофосфамида в режиме профилактики острой РТПХ (69 % против 33 %, р = 0,013). Выполнение алло-ТГСК в срок более 13 мес от постановки диагноза увеличивает вероятность развития острой РТПХ (80 % против 43 %, р = 0,014). Хроническая РТПХ зарегистрирована у 10 пациентов, но лишь 2 ребенка имели распространенную форму, требующую назначения системной̆ терапии. У всех пациентов, достигших приживления трансплантата, нормализация уровня фермента регистрировалась уже в первой точке контроля в промежутке между Д+30 и Д+60 и далее сохранялась в диапазоне нормальных значений, в связи с чем ФЗТ прекращали. При отторжении трансплантата, подтвержденном снижением уровня альфа-L-идуронидазы, ФЗТ возобновляли до выполнения повторной алло-ТГСК. Также оценивали динамику экскреции ГАГ с мочой (рис. 2б), где после алло-ТГСК зарегистрировано плавное снижение показателя до нормальных значений. Основной фактор, определяющий успешность алло-ТГСК у пациентов с МПС I-Н, — восстановление интеллектуального развития детей. Тестирование когнитивных функций было проведено у 18 пациентов (срок после алло-ТГСК превышал 12 мес). Когнитивные функции оценивали посредством анализа показателей внимания и концентрации (методика «10 предметов»), объема словарного запаса (методика речевых игр), умения считать, выполнять инструкции, абстрактного мышления (методика ассоциативных рядов), способности к умозаключениям (тесты на словесно-логическое мышление). До алло-ТГСК все протестированные пациенты имели темповую задержку развития, а также отсутствие речевых навыков. На момент выполнения анализа 4 пациента, достигшие школьного возраста, посещали общеобразовательную школу, 10 детей младшего возраста посещали учреждения дошкольного образования общего типа. В таблице представлены результаты оценки основных когнитивных функций. Возможность сохранения когнитивных способностей существует и в случаях, когда алло-ТГСК выполнена в более позднем возрасте по сравнению с рекомендованными сроками. Все пациенты в последующем находились под наблюдением врачей, осуществлявших коррекцию имевшихся до алло-ТГСК нарушений (окулисты, ортопеды, ЛОР и др.), реабилитологов.
Синдром Гурлер — заболевание с вариабельной клинической симптоматикой и прогрессирующим характером, ведущее к инвалидизации. В первые месяцы жизни дети зачастую длительно наблюдаются у врачей различных специальностей с изолированными диагнозами, что затягивает своевременную постановку основного диагноза. Комплексный подход к наблюдению подобных пациентов позволяет своевременно распознать заболевание и определить тактику дальнейшей терапии. Необходимо отметить, что полиморфизм клинических проявлений МПС 1-Н требует внимательного подхода на этапе диагностики. Это снижает вероятность ошибочного диагноза, уменьшает сроки его установления. Ранняя диагностика способствует своевременности выполнения алло-ТГСК у детей с синдромом Гурлер.
Читайте также
- 30 лет первой аллогенной трансплантации гемопоэтических стволовых клеток у ребенка в России
- Глиомы у детей до года: новейшие методы терапии
- Опухолевый тромбоз нижней полой вены с распространением в правое предсердие
- Современные возможности лучевой визуализации
- Неважно, сколько дней в твоей жизни. Важно, сколько жизни в твоих днях!
- Роль педиатра в успешном лечении гемобластозов у детей
- Адоптивная клеточная иммунотерапия солидных опухолей у детей
- Клинический пример терапии ингибитором рецептора интерлейкина‑1 — препаратом анакинра — у пациента с криопирин-ассоциированным периодическим синдромом
НАШИ ПАРТНЕРЫ















